文章标题
国际SMA关爱日
关爱SMA,JAX小鼠助力患者走向希望
关爱SMA,JAX小鼠助力患者走向希望
作者:陈汀蕙 博士 2023年8月7日
文章内容
奔跑、行走、吞咽、呼吸,这些我们习以为常的“本能”,却是有些人梦寐以求的事情。有这样一种疾病,由于编码运动神经元存活蛋白(Survival motor neuron protein, SMN)的SMN1基因第7号外显子缺失,导致患者出现进行性的肌无力和肌萎缩,甚至危及生命——这便是脊髓性肌萎缩症(Spinal muscular atrophy, SMA)。自1996年起,每年的8月定为全球SMA关爱月;2018年中国SMA患者组织建议,将每年的8月7日设立为“国际SMA关爱日”。在这个特殊的日子,让我们一起了解这种疾病,以及在药物研发中做出重要贡献的动物模型。
全球范围内,SMA发病率为1/10000,是一种罕见病;但在正常人群中,SMA致病基因的携带率非常高,平均每50个普通人中就有一个是携带者1。虽然高达94%的SMA患者携带SMN1基因突变,SMA的严重程度不完全相同,这是因为SMN2基因的拷贝数不同。SMN2基因也能编码SMN蛋白,但由于同义点突变,在进行蛋白质翻译的时候有90%的概率跳过第7号外显子,产生不稳定的截短型蛋白,从而无法发挥SMN蛋白的生理作用。由于小鼠只具有人SMN1基因的同源基因Smn,因此往往采用转基因的方式构建动物模型,使小鼠表达人源SMN基因,以更好模拟人类中SMN蛋白的合成过程。
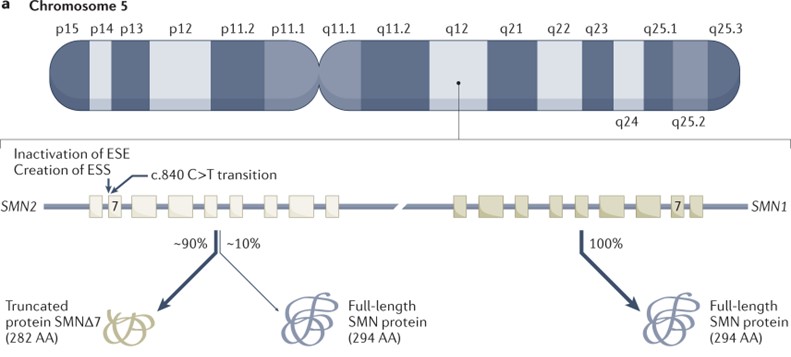
SMN1 及 SMN2 基因示意图1
基于发病年龄和未经治疗时患者能达到的最大运动功能,SMA被分为0-Ⅳ共5种类型。最常见的为Ⅰ型SMA:未经治疗的患儿即使在有辅助的情况下也无法达到坐着时所需要的肌力,通常于2岁前死亡2。目前,FDA已批准3种药物用于治疗SMA3。下面我们将按药物类型进行简要介绍,并带大家一同了解用于药物研发的动物模型。
基因替代疗法
2019年5月,美国食品药品监督管理局(Food and Drug Administration, FDA)正式批准由Novartis公司研发的Onasemnogene abeparvovec(商品名Zolgensma)上市,用于治疗2岁以下的SMA患儿4。这种基因疗法利用scAAV9载体,将正常SMN1基因导入患者体内,从而改善运动神经元等受累细胞的功能。在该药物的研发过程中,使用了猕猴和FVB.Cg-Grm7Tg(SMN2)89Ahmb Smn1tm1Msd Tg(SMN2*delta7)4299Ahmb/J (#005025)小鼠(通常称为FVB.SMNΔ7;SMN2;Smn-小鼠)。FVB.SMNΔ7;SMN2;Smn-小鼠携带两个转基因等位基因和一个无效突变,缺乏内源性Smn基因的同时表达人源SMN2基因,以及缺乏7号外显子的人源SMN2 cDNA。在出生后,该品系小鼠的体型明显小于正常幼崽,在出生后5天内表现出肌无力的现象,并随着年龄增加逐渐加剧。该品系小鼠14日龄时可检测到腓肠肌萎缩,可以较好的模拟SMA患者的神经病理表现和症状,最常用于与早期干预相关的疗效研究。在Onasemnogene abeparvovec的临床前研究中,出生后1天时给予SMA小鼠心内注射AAV,可以挽救其运动功能、神经肌肉电生理活动,并延长寿命5。
反义寡核苷酸(ASO)
由Biogen公司生产的Nusinersen(商品名Spinraza)是首个获得FDA批准的用于治疗SMA的药物。这是一种ASO药物,通过鞘内给药,Nusinersen可以与SMN2基因7号外显子的剪切位点结合,降低其在剪切过程中被截短的比例,从而增加全长SMN蛋白的合成6。在该药物的研发过程中,同样使用了猕猴,以及另一种SMA模型小鼠——FVB.Cg-Smn1tm1Hung Tg(SMN2)2Hung/J (#005058)小鼠。该品系小鼠携带Smn1tm1Hung敲除等位基因,同时转入整个人源SMN2基因编码区及侧翼序列。该品系半合子小鼠携带约2个转基因拷贝,并且小鼠转基因的拷贝数与神经退行性表型的严重程度之间有很强的相关性:双纯合子小鼠表现出Ⅲ型SMA的症状,可出现脊髓前角运动大神经元的丢失;而Smn1tm1Hung纯合Tg(SMN2)2Hung转基因半合小鼠则表现出Ⅰ型SMA的症状,通常在约14天时死亡。研究者向新生鼠或成年小鼠侧脑室注射Nusinersen后,可观察到小鼠耳朵和尾部坏死情况有所改善6。并且,成年小鼠相关研究结果显示,该ASO药物停药后6个月仍能观察到药物的最大效应7。
小分子药物
由PTC Therapeutics、非营利性SMA基金会和Roche公司三方联合开发的Risdiplam (商品名Evrysdi),因其便捷的口服给药方式而受到大家的关注。Risdiplam是一种高效的SMN2剪接修饰剂,它可以稳定7号外显子5’端剪接位点与剪接体中U1小核核糖核蛋白(small nuclear ribonucleoprotein, snRNP)之间的相互作用,充当分子间的“胶水”,从而修饰剪接8。在Risdiplam的临床前研究中使用了FVB.SMNΔ7;SMN2;Smn-小鼠(#005025)和FVB.129(B6)-Smn1tm5(Smn1/SMN2)Mrph/J (#008604)小鼠(通常称为Smn1C小鼠)。Smn1C小鼠含有两个串联的Smn1/SMN2杂交基因。由于7号外显子来源于人SMN2基因,产生的Δ7-SMN基因产物比全长SMN产物优先剪接,Smn1C/C小鼠表现出相对较轻的SMA表型。研究者发现,当口服Risdiplam后,两种SMA小鼠模型的全长SMN蛋白水平得到恢复9。同时,SMA模型小鼠的生存时间和体重均得到了不同程度的恢复。
碱基编辑
除了上述三种已获批药物,碱基编辑技术在治疗SMA领域的应用也备受瞩目。2016年,刘如谦(David R. Liu)教授团队开发了该项技术:依托于CRISPR的DNA定位能力,催化特定位置的C或A进行脱氨反应,转变为U或I,然后在DNA复制的过程中被当作T或G,从而实现C到T或A到G的转换。SMN1基因与SMN2基因的差别在于7号外显子第6位核苷酸处有C·G到T·A的转变,导致mRNA中的7号外显子跳跃,产生SMNΔ7截短蛋白。理论上,通过将SMN2基因中的T·A碱基对转换为C·G对,能够有效地将其转化为SMN1基因的副本,从而治疗疾病。体外研究表明,这种方式能有效增加SMN蛋白水平。研究者利用双AAV载体,将腺嘌呤碱基编辑器(Adenine base editors, ABE)导入FVB.SMNΔ7;SMN2;Smn-小鼠(#005025)中枢神经系统。在进行治疗后,FVB.SMNΔ7;SMN2;Smn-小鼠(#005025)的运动功能、存活率及寿命均有所改善10。
目前,JAX可提供多种用于SMA研究的小鼠品系,活体品系如下:
| 用途 | 常用名 | 品系标准命名 | JAX品系号 |
|---|---|---|---|
|
SMA模型鼠 |
SMA-like | FVB.Cg-Smn1tm1Hung Tg(SMN2)2Hung/J | 005058 |
|
FVB.SMNΔ7; SMN2;Smn- |
FVB.Cg-Grm7Tg(SMN2)89Ahmb Smn1tm1Msd Tg(SMN2*delta7)4299Ahmb/J | 005025 | |
| 对照鼠 | Smn delta 7 | FVB.129P2(B6)-Smn1tm1Hung/J | 031678 |
*对照鼠:指FVB.Cg-Smn1tm1Hung Tg(SMN2)2Hung/J (#005058)的对照品系
若您对SMA疾病研究及药物研发感兴趣,可参看品系查询,寻找适合的动物模型。同时,杰克森实验室也提供SMA体内药效研究服务,可协助您检测各种SMA相关指标,包括体重、存活率、电生理活性、神经肌肉接头成熟度以及SMN水平等。您可以查看目前JAX提供的神经生物学药效服务,也可以联系杰克森实验室技术支持(micetech@jax.org.cn, 400-001-2626)获取更多相关信息,我们将竭尽所能为您提供帮助。
参考文献
- Mercuri E et al. “Spinal muscular atrophy”. Nature Reviews Disease Primers. 2022 Aug 4;8(1):52. doi: 10.1038/s41572-022-00380-8. PMID: 35927425.
- Wirth B. “Spinal Muscular Atrophy: In the Challenge Lies a Solution”. Trends in Neurosciences. 2021 Apr;44(4):306-322. doi: 10.1016/j.tins.2020.11.009. Epub 2021 Jan 7. PMID: 33423791.
- Chaytow H et al. “Spinal muscular atrophy: From approved therapies to future therapeutic targets for personalized medicine”. Cell Reports Medicine. 2021 Jul 21;2(7):100346. doi: 10.1016/j.xcrm.2021.100346. PMID: 34337562; PMCID: PMC8324491.
- ZOLGENSMA | FDA
- Foust KD, Wang X, McGovern VL, Braun L, Bevan AK, Haidet AM, Le TT, Morales PR, Rich MM, Burghes AH, Kaspar BK. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010 Mar;28(3):271-4. doi: 10.1038/nbt.1610. Epub 2010 Feb 28. Retraction in: Nat Biotechnol. 2022 Nov;40(11):1692. PMID: 20190738; PMCID: PMC2889698.
- Hua Y et al. “Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model”. Genes & Development. 2010 Aug 1;24(15):1634-44. doi: 10.1101/gad.1941310. Epub 2010 Jul 12. PMID: 20624852; PMCID: PMC2912561.
- Rigo F et al. “Pharmacology of a central nervous system delivered 2'-O-methoxyethyl-modified survival of motor neuron splicing oligonucleotide in mice and nonhuman primates”. The Journal of Pharmacology and Experimental Therapeutics. 2014 Jul;350(1):46-55. doi: 10.1124/jpet.113.212407. Epub 2014 Apr 30. PMID: 24784568; PMCID: PMC4056267.
- Sheridan C. “First small-molecule drug targeting RNA gains momentum”. Nature Biotechnology. 2021 Jan;39(1):6-8. doi: 10.1038/s41587-020-00788-1. Erratum in: Nat Biotechnol. 2021 Feb 4;: PMID: 33432225.
- Ratni H et al. “Specific Correction of Alternative Survival Motor Neuron 2 Splicing by Small Molecules: Discovery of a Potential Novel Medicine To Treat Spinal Muscular Atrophy”. J Med Chem. 2016 Jul 14;59(13):6086-100. doi: 10.1021/acs.jmedchem.6b00459. Epub 2016 Jul 6. PMID: 27299419.
- Arbab M et al. “Base editing rescue of spinal muscular atrophy in cells and in mice”. Science. 2023 Apr 21;380(6642):eadg6518. doi:1126/science.adg6518. Epub 2023 Apr 14. PMID: 36996170; PMCID: PMC10270003.
搜索分组
