舞蹈,在普通人的印象中应当是优美的、欢快的,然而,却有一种叫做亨廷顿舞蹈症的疾病,颠覆着人们对于“舞蹈”的认知。
2.7/100000
亨廷顿舞蹈症是一种罕见的、常染色体单基因显性遗传病,全球患病率约为十万分之2.7。患者多在35~50岁发病,表现出不自主舞蹈样震颤、精神障碍、痴呆。起初症状和体征较为隐匿,随后呈缓慢进行性加重,平均生存期为10-20年,目前无药可医。
每年的5月15日是国际亨廷顿舞蹈症关爱日,让我们一起关注这一疾病,同时也共同努力,争取早日攻克这一医学难题。
发病机制
亨廷顿舞蹈症是由于四号染色体上的亨廷顿基因(HTT)中编码谷氨酸的CAG重复序列拷贝数异常增多,产生了突变的亨廷顿蛋白(mHTT),在神经元内积累,影响神经细胞的正常功能,最终导致疾病。正常人群CAG重复数小于26,当超过36则被称为突变型,CAG拷贝数愈多,疾病发生的年龄愈早,后果也愈严重。
亨廷顿舞蹈症的诊断依靠典型症状、体征和阳性家族史,确诊需进行基因检测CAG重复数。
| CAG 重复数 | 是否携带HD致病基因 | 临床表现 |
| ≤26 | 否 | 正常 |
| 27-35 | 减数分裂时CAG重复数会发生扩展突变 | 正常,但后代可能是患者 |
| 36-39 | 携带不完全外显的HD等位基因 | 可能发病也可能不发病 |
| ≥40 | 携带完全外显的HD等位基因 | 患者 |
治疗手段
目前,亨廷顿舞蹈症的治疗主要是支持治疗和对症治疗,来缓解患者的震颤(VMAT2抑制剂、安定类药物)和精神障碍(SSRI类、SNRI类药物),改善病人生活质量,但仍无法根治或改变疾病的发展进程,研究人员仍在继续寻找特异性治疗方法。
多种基因疗法,例如靶向突变型亨廷顿蛋白mRNA的反义寡核苷酸(ASO)疗法[1]、miRNA疗法[2]、CRISPR-Cas9基因编辑[3, 8]、siRNA疗法[4]、CRISPR-Cas13d基因编辑技术[5]等在动物模型上取得了显著成效,目前也有几款药物也进入了临床阶段,包括三款ASO疗法(Roche/Ionis的Tominersen,Wave Life Sciences公司的WVE-003和VICO Therapeutics BV公司的VO-659)和两款AAV载体介导的基因疗法(uniQure公司的AMT-130和Asklepios公司的BV-101)。这些疗法目前都还处于临床早期阶段,其最终疗效如何还有待进一步验证。
小鼠模型
亨廷顿(HD)小鼠模型可以模拟病人的疾病表型及病理特征,为发病机制研究和开发有效的治疗方法提供了关键信息。上述进入临床阶段的药物,其临床前阶段都在小鼠模型上进行过筛选和验证。JAX有70多种HD小鼠模型,大致可以分为以下三类:
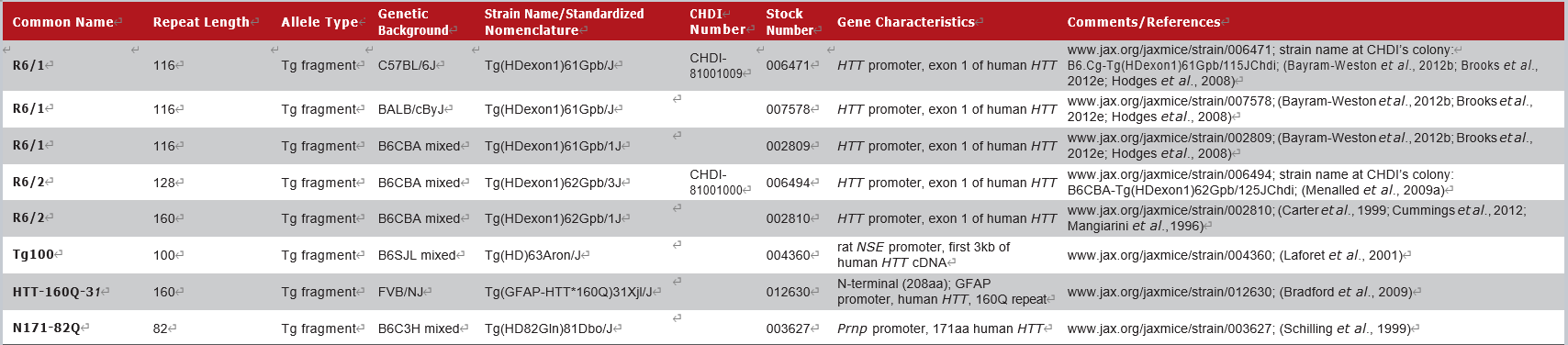
1 HTT的N端转基因模型
这一类模型携带了人类HTT基因5'端的一小部分,包括含有CAG重复区exon 1。通常呈现加速表型[6],并出现进行性的神经系统异常,包括失去协调性、震颤、运动不足、步态异常、神经病理学和过早死亡。
R6/1和R6/2小鼠表达突变的人类HTT exon 1,是第一个转基因品系,也是最广泛使用的HD小鼠模型。Cas9疗法的概念验证[3],siRNA疗法的验证[4],以及Asklepios公司的基因疗法BV-101的临床前实验[7],就是在R6/2小鼠(002810)体内进行的。
JAX拥有的部分HTT-N端转基因小鼠模型如下:
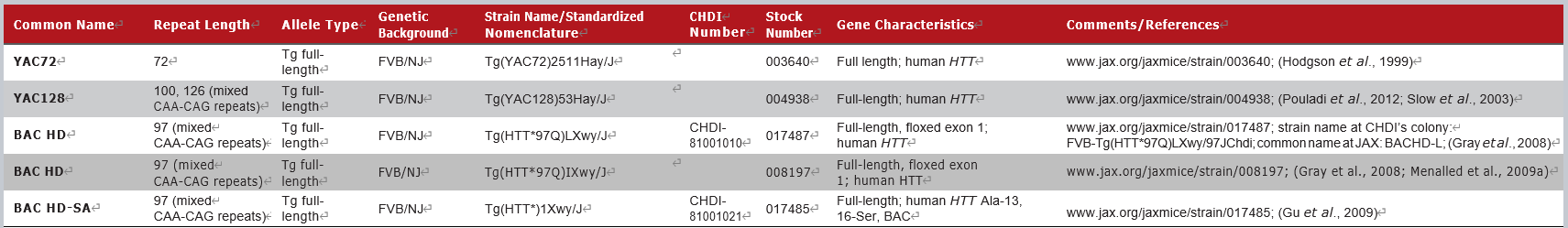
2 HTT全长转基因模型
这一类模型通过酵母或细菌人工染色体(YAC或BAC)的方法表达人源的全长突变亨廷顿蛋白。与N端转基因模型不同,这类模型通常发病更晚,具有较慢的、进行性的认知和运动障碍,并显示出相对正常的存活率。但这类模型在测试直接针对人类HTT基因或蛋白的实验性疗法时更具有优势。例如ASO疗法[1]和siRNA疗法[4]都曾在BAC HD小鼠(008197)和YAC128小鼠(004938)上进行过验证。
JAX拥有的部分HTT全长转基因小鼠模型如下:
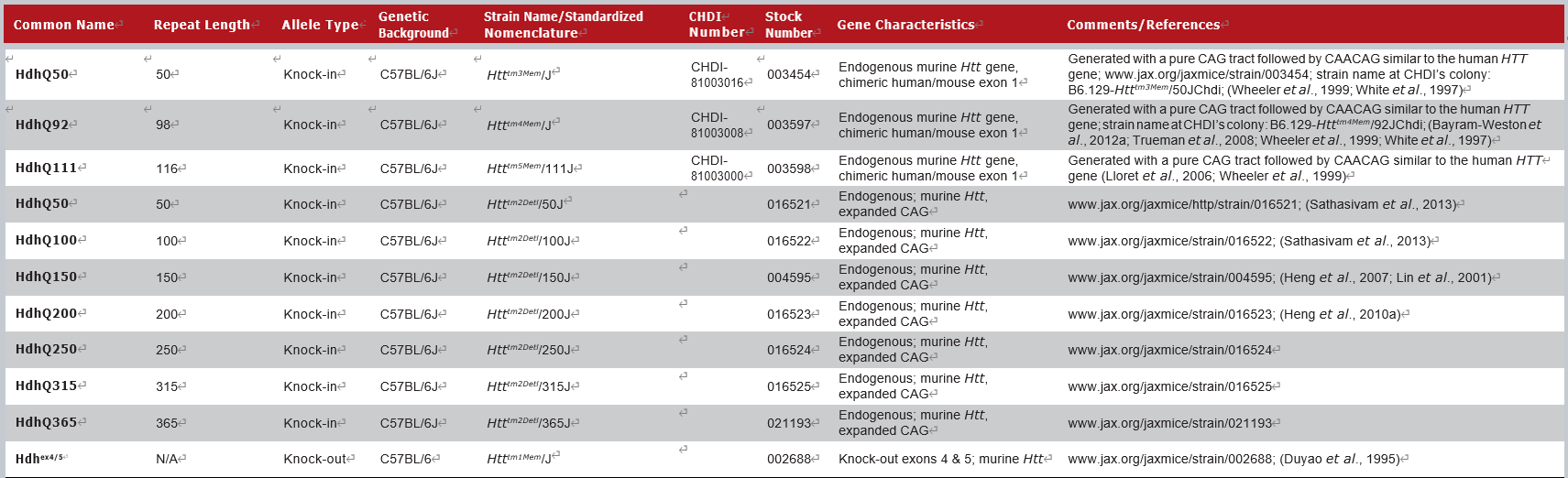
3 Knock-in模型
这一类模型是通过同源重组的方式,直接将小鼠的Htt基因exon1替换为人HTT的CAG重复序列,利用小鼠内源Htt启动子表达HTT蛋白。这类模型可以精确控制CAG的重复数目和HTT的表达水平,可以构建多种不同CAG重复数的品系,用于研究CAG重复数的功能。按照不同的CAG重复数,常用的小鼠模型有Q140、Q150、Q175等几种。CRISPR/Cas9基因疗法也在Q140小鼠模型上进行了验证[8]。
JAX拥有的部分Knock-in小鼠模型如下:
更多HD小鼠模型的资料,可以联系杰克森实验室技术支持(micetech@jax.org.cn, 400-001-2626)获取。
[1] Kordasiewicz, Holly B et al. “Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis.” Neuron vol. 74,6(2012):1031-44.doi:10.1016/j.neuron.2012.05.009
[2] Pfister, Edith L et al. “Artificial miRNAs Reduce Human Mutant Huntingtin Throughout the Striatum in a Transgenic Sheep Model of Huntington's Disease.” Human gene therapy vol. 29,6 (2018): 663-673. doi:10.1089/hum.2017.199
[3] Ekman, Freja K et al. “CRISPR-Cas9-Mediated Genome Editing Increases Lifespan and Improves Motor Deficits in a Huntington's Disease Mouse Model.” Molecular therapy. Nucleic acids vol. 17 (2019): 829-839. doi:10.1016/j.omtn.2019.07.009
[4] Zhang, Li et al. “Therapeutic reversal of Huntington's disease by in vivo self-assembled siRNAs.” Brain : a journal of neurology vol. 144,11 (2021): 3421-3435. doi:10.1093/brain/awab354
[5] Morelli, Kathryn H et al. “An RNA-targeting CRISPR-Cas13d system alleviates disease-related phenotypes in Huntington's disease models.” Nature neuroscience vol. 26,1 (2023): 27-38. doi:10.1038/s41593-022-01207-1
[6] Menalled, Liliana et al. “Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models.” Neurobiology of disease vol. 35,3 (2009): 319-36. doi:10.1016/j.nbd.2009.05.007
[7] Boussicault, Lydie et al. “CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington's disease.” Brain : a journal of neurology vol. 139,Pt 3 (2016): 953-70. doi:10.1093/brain/awv384
[8] Yang, Su et al. “CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease.” The Journal of clinical investigation vol. 127,7 (2017): 2719-2724. doi:10.1172/JCI92087
